α-芳基乙腈作为一种常见的结构单元在药物化学的研究中具有颇为广泛的应用,尤其是α位为季碳时,相应位点在代谢过程中无法进一步氧化,极大地避免了CN-的释放,由此使α-芳基乙腈发挥其生理活性的同时又能确保给药的安全性。目前,不少药物分子中均包含这类结构,如应对晚期乳腺癌的阿那曲唑(anastrazole),治疗心绞痛、高血压的维拉帕米(verapamil)。除此之外,α-芳基乙腈还可通过适当的化学转化形成相应的羧酸、醛/酮、酰胺等不同类型的化合物,因而在有机合成领域亦用作重要的合成砌块。

部分包含α-芳基乙腈结构的药物分子(图片来源:参考资料[1])
早期构建α-芳基乙腈主要从(拟)卤化苄出发,使用亲核氰基化试剂(NaCN、KCN等)对其亲核取代来完成。但体系一般需加热至较高的温度,并且反应效率不高,加之还需要使用毒性强的氰化物,为该类化合物的合成带来了重重阻碍。另一种方法则是首先制备相应的芳香酰胺,在P2O5、SOCl2等脱水剂的作用下脱水得到目标产物。这些脱水剂酸性较强,意味着对酸敏感的官能团无法兼容,底物的适用范围会受到一定限制。
还有一种可行的方案是以乙腈及其衍生物作为原料,种类丰富、易得的(拟)卤代芳香烃作为亲电试剂对其α-芳基化来合成α-芳基乙腈。这种方法无需预先制备相应的芳香酰胺,同时也不涉及氰化物的使用,简单快捷,也更为安全,但直接将两者偶联较为困难。从反应机制上讲,以上过程属于芳香亲核取代(SNAr),参与反应的(拟)卤代芳香烃需同时修饰强吸电子基团(如NO2)进行活化。
2000年,美国辉瑞(Pfizer)公司的Stéphane Caron博士等人对反应条件进行了改进,此时氟代苯作为芳基化来源无需修饰吸电子取代基,富电子的底物甚至也可顺利参与反应。反应中的碱KHMDS起到了关键的作用,腈的α位去质子化后可与K+结合形成烯亚氨基钾加合物,K+具有一定的π酸性,可同时与氟代苯的苯环作用,由此发挥活化底物的功效,并推动后续的亲核加成、取代进行。不过,该方法仅适用于亲核性较强的二级腈,一级腈或修饰其他吸电子基团的二级腈(如氰乙酸乙酯)参与反应的效果较差。
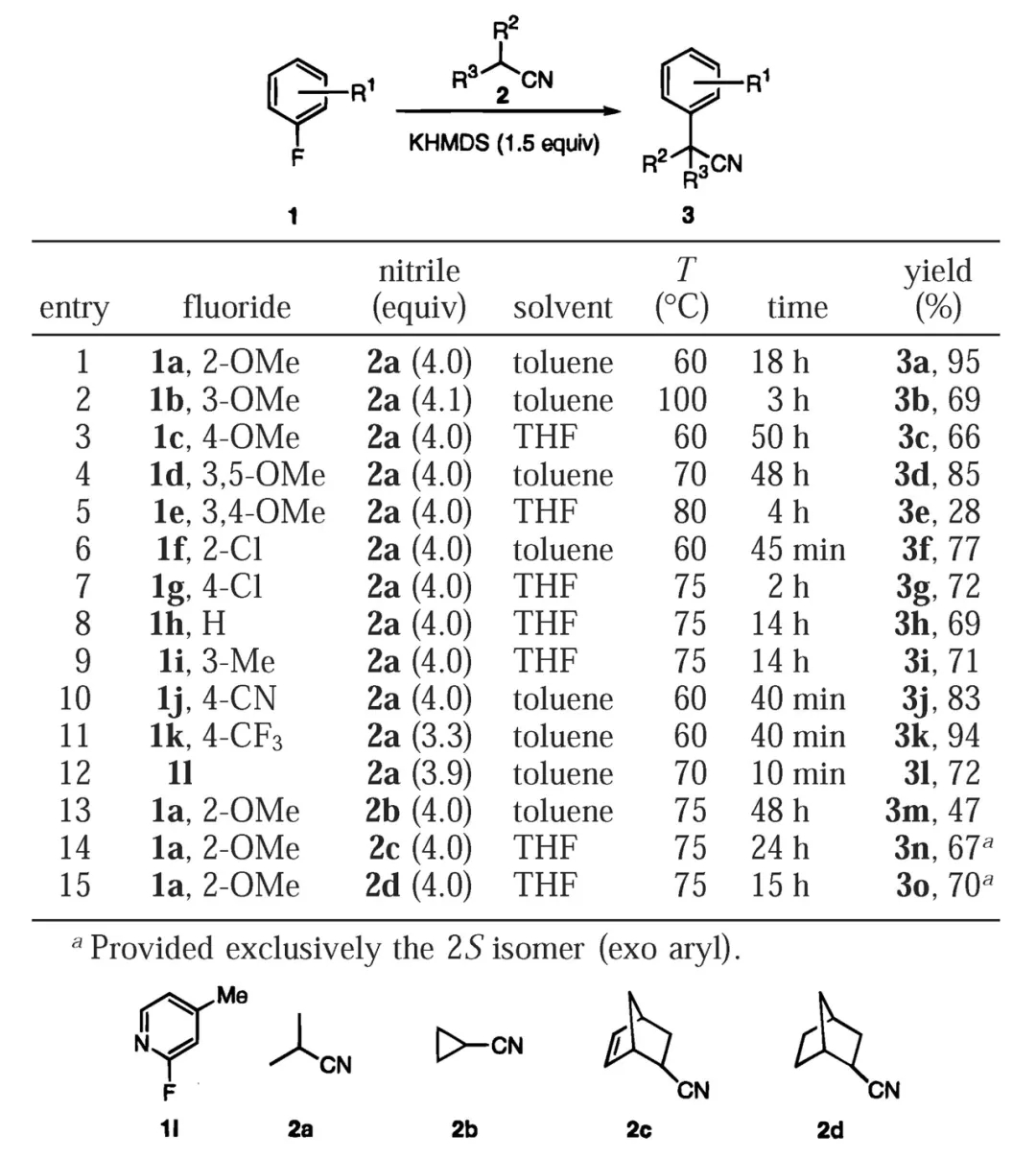
氟代苯与二级腈反应合成α-芳基乙腈(图片来源:参考资料[2])
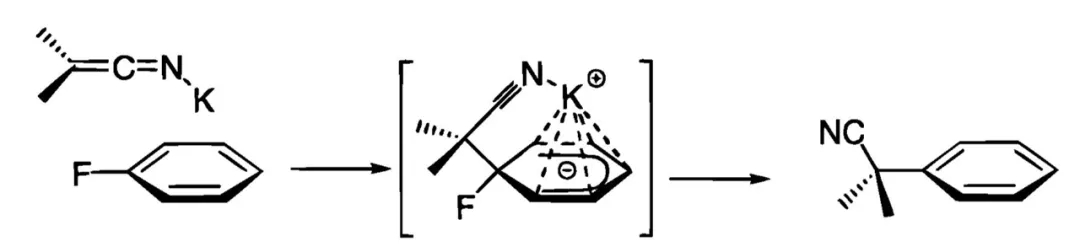
二级腈对氟代苯芳香亲核取代可能的反应机制(图片来源:参考资料[2])
而随着过渡金属催化反应的发展,人们同样开始考虑借助合适的过渡金属催化剂来完成乙腈及其衍生物与(拟)卤代芳香烃的C-C键偶联。今天我们将为大家介绍一下中科院上海有机化学研究所马大为院士团队在该研究领域取得的进展,他们利用此前发展的Cu/草酰二胺催化体系,实现了氯/溴代(杂)芳香烃与氰乙酸乙酯的Ullmann型高效偶联。不仅如此,氰化试剂还可进一步拓展至α-烷基取代的氰乙酸乙酯,相关工作发表在化学期刊Angew. Chem. Int. Ed.上。在详细讨论这项工作之前,我们不妨来了解一下过渡金属催化剂参与合成α-芳基乙腈研究的大体情况。

图片来源:参考资料[3]
目前,用于构建α-芳基乙腈的过渡金属催化体系主要分为Pd和Cu两类。前者最早由时任美国耶鲁大学(Yale University)教授的John F. Hartwig建立。2002年,该团队合成了几种膦配体配位的氰烷基芳香钯配合物,并对其结构及还原消除的活性进行了探究,由此设计出Pd(OAc)2/BINAP、Pd2dba3·CHCl3/PtBu3两种Pd催化体系。这两种组合均可实现烷基腈与溴代苯的C-C键偶联,对于后者,无论是富电子还是贫电子底物,均可顺利发生反应。但与此前Stéphane Caron博士的工作类似,腈的适用性仍需进一步改善,二级腈、苯乙腈参与C-C键偶联的效果较好,但其他一级腈(如乙腈、正丁腈)会出现α-双芳基化,可能是因为形成的α-芳基乙腈可进一步去质子化,并且很容易与Pd催化剂配位再次反应。另外,反应仍旧需要使用强碱LiHMDS、NaHMDS,底物中对碱敏感的官能团会受到明显的影响。

John F. Hartwig教授早期建立的Pd催化体系(图片来源:参考资料[4])
第二年,美国爱荷华州立大学(Iowa State University)的John G. Verkade教授则巧妙利用该团队发展的含磷双环有机非离子超强碱(Verkade碱)作为配体,与Pd(OAc)2或Pd2dba3结合,可实现反应活性更低的氯代芳香烃与烷基腈的偶联。此时氰乙酸乙酯也可作为氰化来源高效参与反应,但一级腈发生过度α-芳基化的问题仍旧未解决。

Verkade碱作为配体实现Pd催化氯代芳香烃与烷基腈的偶联(图片来源:参考资料[5])
为此,John F. Hartwig教授将烷基腈预先转化为相应的α-三甲硅基腈,并加入ZnF2,反应过程中可形成氰烷基锌中间体,进而通过Negishi型C-C键偶联实现乙腈等一级腈的选择性α-单芳基化。α-双芳基化产物得到有效的抑制,并且由于体系中不再加入强碱,官能团兼容性也得以改善。
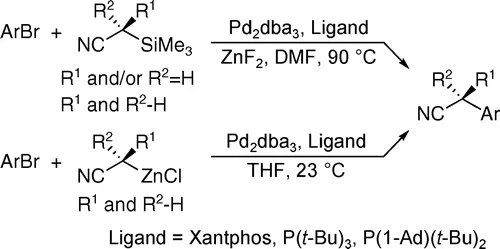
α-三甲硅基腈作为氰化试剂合成α-芳基乙腈(图片来源:参考资料[6])
其他研究团队也相继尝试了各种不同类型的氰化试剂。2011年,清华大学的刘磊教授团队还使用氰乙酸盐作为原料,与(拟)卤代芳香烃通过Pd催化的脱羧偶联完成了α-芳基乙腈的合成,单芳基化的选择性同样十分出色。同年,德国科隆大学(University of Cologne)的Hans-Günther Schmalz教授还与瑞士诺华(Novartis)公司的Juraj Velcicky合作,以异噁唑作为氰亚甲基前体,使用异噁唑硼酸频哪醇酯与卤代芳香烃进行Suzuki偶联形成芳基异噁唑,经后续转化得到α-芳基乙腈。不过,无论对于哪种反应体系,大多数卤代杂芳香烃参与反应的效果仍不够理想。
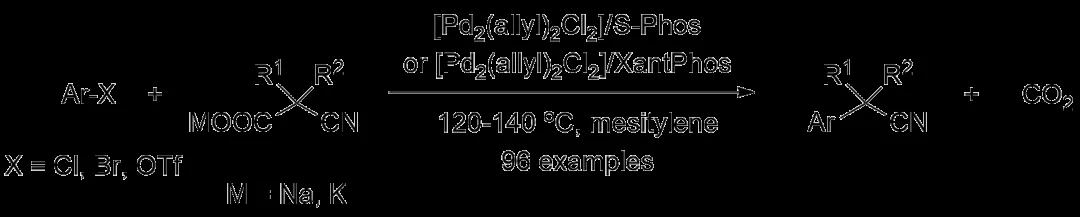
氰乙酸盐作为氰化来源合成α-芳基乙腈(图片来源:参考资料[7])

噁唑硼酸频哪醇酯作为氰亚甲基前体合成α-芳基乙腈(图片来源:参考资料[8])
Cu催化构建α-芳基乙腈的工作其实出现得更早。1983年,日本爱媛大学(Ehime University)的Atsuhiro Osuka教授便以CuI作为催化剂,设计了碘代苯与氰乙酸乙酯钠盐的偶联过程,随后酯基水解并脱羧便可得到相应的α-芳基乙腈。后续人们也对上述反应体系进行了细微调整,但对于芳基化来源,长期以来仅活性较高的碘代芳香烃参与反应的效果较好,氯、溴代芳香烃则不尽如人意。马大为院士团队在2015年设计了一类草酰二胺配体,与Cu催化剂结合可以完成氯代苯与胺的高效Ullmann偶联,氯代杂芳香烃也可以顺利参与反应。后续他还将这种Cu/草酰二胺催化体系用于卤代(杂)芳香烃与氮杂芳香烃、酰胺等含氮试剂的C-N键偶联,甚至还能借助酚、醇等含氧来源构建C-O键,均取得了不错的结果。
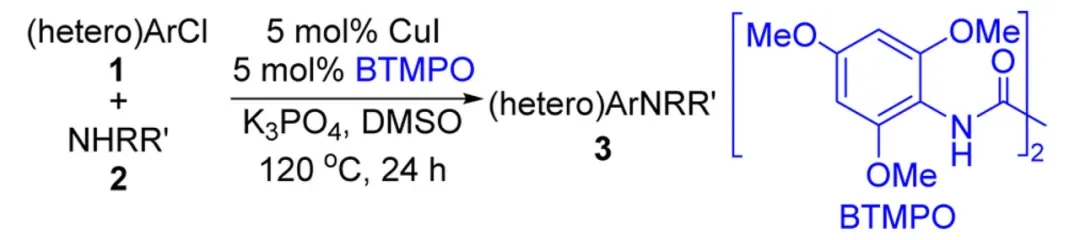
草酰二胺类配体BTMPO与CuI结合实现氯代(杂)芳香烃与胺的Ullmann偶联(图片来源:参考资料[10])
于是,该团队设想进一步利用这种催化体系合成α-芳基乙腈。他们起初以4-氯苯甲醚作为芳基化来源,氰乙酸乙酯作为氰化试剂,考察不同配体与CuX(X = Cl, Br, I)结合用作催化剂时两者发生C-C键偶联的反应情况。有趣的是,草酰二胺配体L1(MNBO)与L2(MNPMO)仅侧链的其中一个芳香环有所不同(前者为苯、后者为吡啶),但用作配体的反应效果却存在明显的差异,可能的原因在于吡啶的吸电子特性对Cu金属中心的电子密度具有一定影响,从而对催化效果产生微妙的作用。最终他们选择CuBr作为预催化剂,L2作为配体,选择碱tBuONa对氰乙酸乙酯去质子化,C-C键偶联过程结束后加入适量的水使酯基水解并脱羧,能以良好的产率得到4-甲氧基苯乙腈产物。

反应条件的优化(图片来源:参考资料[3])
该方法对于不同结构的氯代芳香烃均具有良好的适用性,无论其修饰给电子还是吸电子取代基,反应均可顺利进行。溴代芳香烃也用作芳基化来源,不过反应条件需要细微的调整,其中CuCl用作预催化剂,K3PO4作为碱,此时催化剂负载量更低,反应温度也可进一步降低。相比之下,氯代芳香烃参与C-C键偶联受空间位阻的影响也更大。当然,卤代(杂)芳香烃作为底物也取得了满意的结果。

氯、溴代(杂)芳香烃底物适用范围的考察(图片来源:参考资料[3])
作者还进一步使用α-烷基取代氰乙酸乙酯作为氰化试剂考察其与溴代(杂)芳香烃的反应情况,此时换用配体L5(APMO)可进一步提高反应的效率。这种构建α位同时修饰其他取代基的α-芳基乙腈的方法在药物研发领域具有重要的意义。

α-烷基取代氰乙酸乙酯作为氰化试剂合成α-芳基乙腈(图片来源:参考资料[3])
近年来,马大为院士团队发展的Cu/草酰二胺催化体系在用于构建芳香C-N键、C-O键时取得了切实的应用,相信本文介绍的合成α-芳基乙腈的方法也会在相关研究中拥有不俗的表现。
关键词:芳基乙腈 草酰二胺
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2025 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1






 发询盘
发询盘