- 0
-
 平台客服:前小衍
平台客服:前小衍 联系电话:027-81293128
联系电话:027-81293128 -
您的随身业务助手
微信扫一扫 关注「前衍化学」公众号


 发询盘 看报价收询盘 拿订单
发询盘 看报价收询盘 拿订单
微信扫一扫 关注「前衍化学」公众号
发询盘 看报价收询盘 拿订单2018年12月28日,国家药典委员会发布了关于《中国药典》2020年版四部通则增修订内容(第三批)的公示。


1、元素杂质限度和测定指导原则
根据ICH Q3D新增,预计影响较大。适用于化药,不适用于中药、中成药、放射性药物、疫苗、细胞代谢产物、 DNA 产品、过敏原提取物、细胞、全血、血细胞成份或包括血浆及血浆衍生物在内的血液衍生物、非体循环透析液, 以及为了治疗作用而人为引入到药品中的元素;也不适用于基于基因(基因治疗)、细胞(细胞治疗)和组织(组织工程)的药品。
文件中规定的限度不直接适用于原料药和辅料。但为使药品中的元素杂质限度能符合规定, 药品生产企业需要一些原料药或辅料中元素杂质含量(或浓度) 相关信息。药品生产企业可以使用原料药或辅料生产企业提供的元素杂质测试数据或者风险评估报告。原料药和辅料的合格供应商提供的元素杂质数据可供药品生产企业用于证明最终药品是否符合本指导原则
的限度要求。

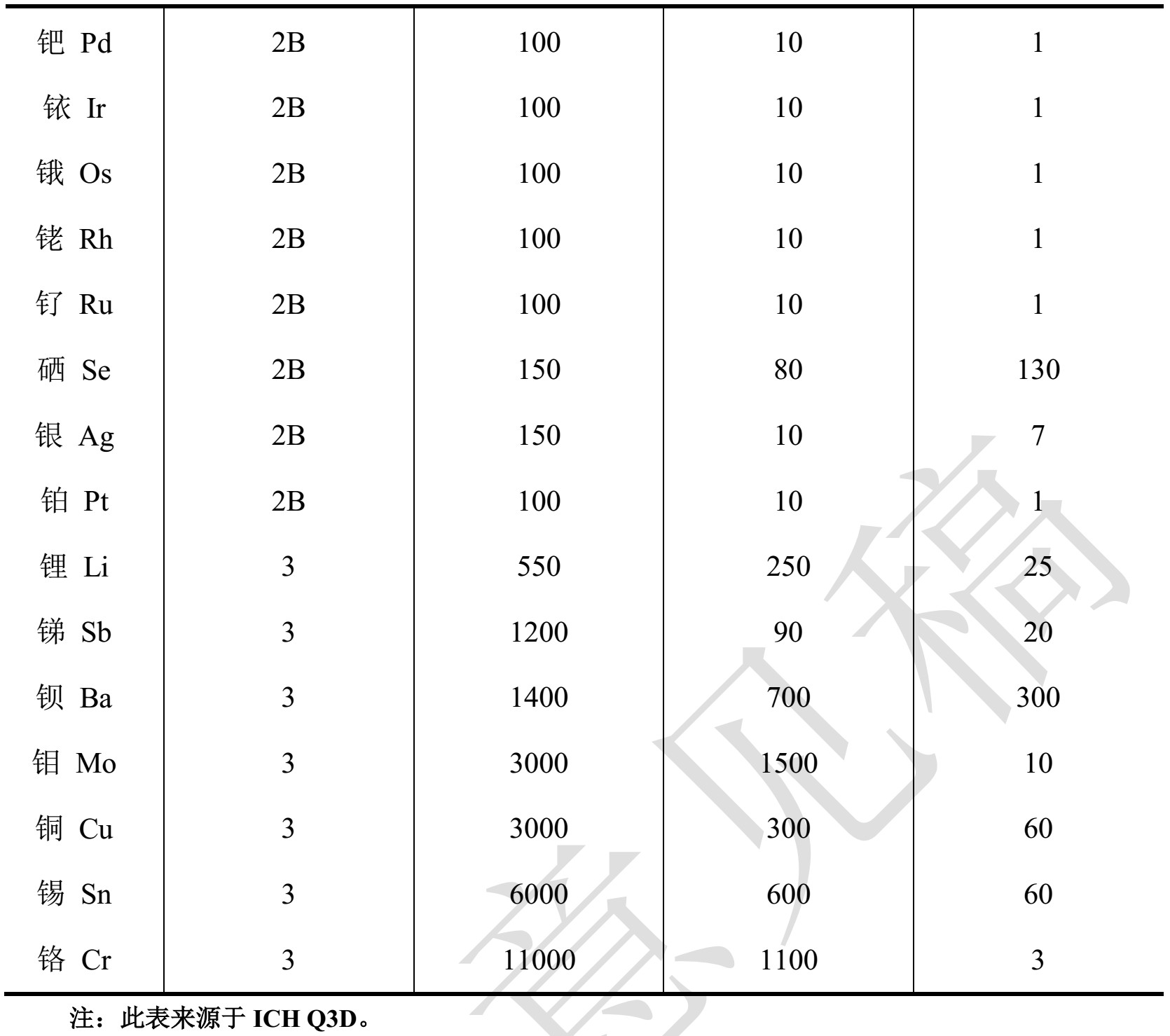
1 类元素是对人体有害元素,在药品生产中禁用或限制使用。
2 类元素通常被认为是给药途径依赖型的人体有害元素。根据它们出现于药品中的相对可能性,进一步分成 2A 和 2B 亚类。
3 类元素口服给药途径的毒性相对较低(高 PDE 值,通常>500µg/天),但在吸入和注射给药途径的风险评估中仍需考虑。
文件中给出详细的限度确认方法:制剂分析法、加和法、单组分法。
文件解释如果药品生产企业通过工艺监测和供应链控制,可以证明并保证制剂符合本指导原则规定的限度要求,则可不必进一步监测。
控制阈值,即PDE 值的 30%,可用于判断药品中的元素杂质是否需要额外的控制。
如果药品中某个元素杂质水平总是小于 PDE 值的 30%,只要对数据进行了适当的评估并表明已对元素杂质进行了足够的控制,则不再需要额外的控制。
如果风险评估无法表明某个元素杂质水平始终低于控制阈值,就需要建立控制方法以保证药品中元素杂质水平不超过 PDE 值。
元素杂质的控制方法包括:
调整相关生产工艺,通过特定或非特定的纯化步骤将元素杂质降低至控制阈值之下;
实施工艺过程的中游或上游控制,将药品中元素杂质的浓度限制在控制阈值以下;
建立辅料或物料(如:合成中间体)的元素杂质标准限度;
建立原料药的元素杂质标准限度;
建立制剂的元素杂质标准限度;
选择合适的包装材料;
对药品中元素杂质进行定期检测。
在第二法(烘干法)中,规定了样品取样量要求2g-5g,取样量增加。增加了样品前处理步骤。规定了两次称重差异不超过5mg为恒重。

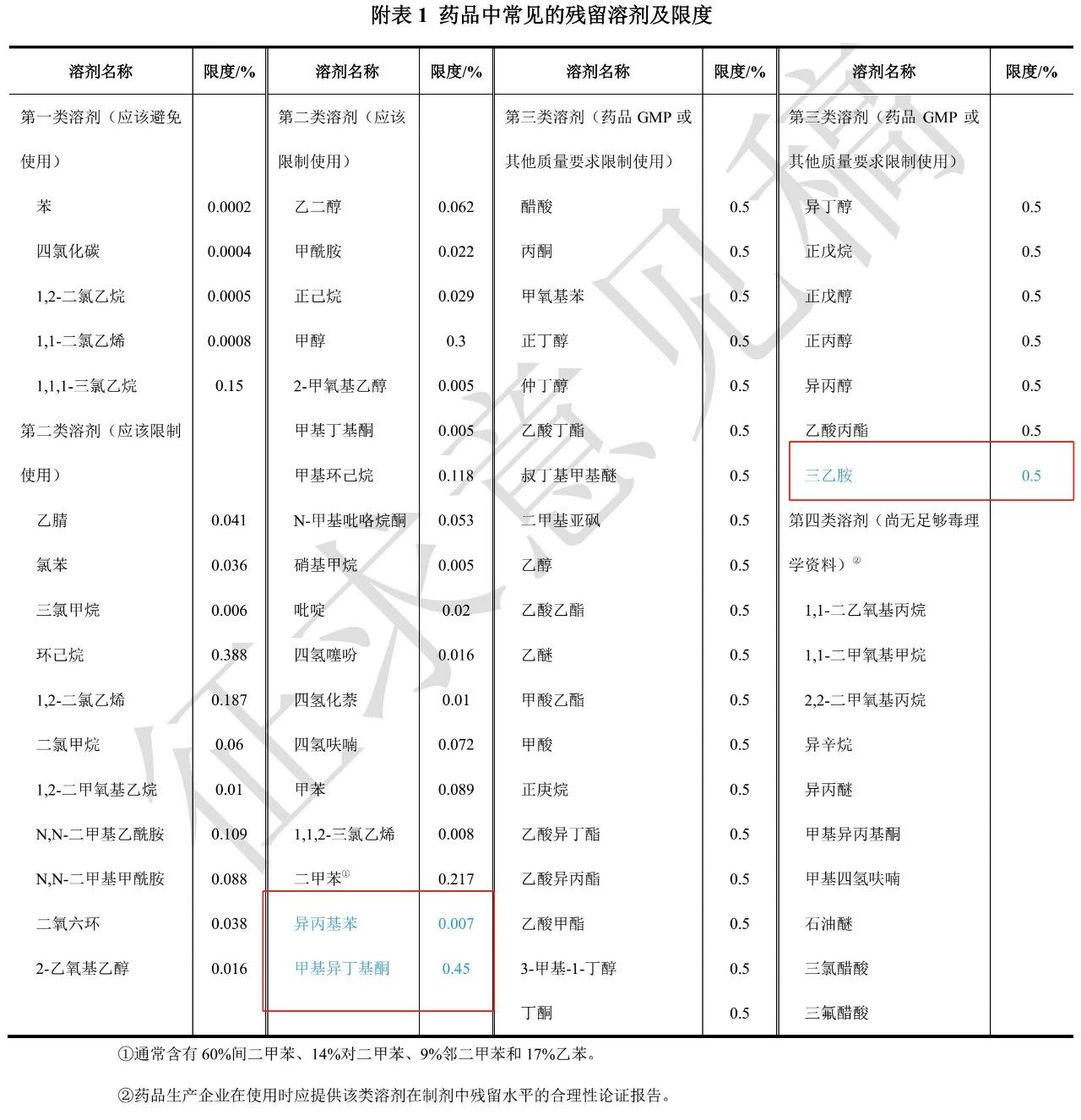
3、残留溶剂测定法
残留溶剂种类及限度变化是一个主要变化,新增了二类溶剂异丙基苯0.007%、甲基异丁基酮0.45%和三类溶剂三乙胺0.5%(原EMA规定为0.032%),需要引起注意。这些充分借鉴了ICHQ3C的最新版本。
4、分析方法验证指导原则
分析方法验证指导原则中变化很大的是删除了关于“校正因子”的验证内容。


对于可接受限度的表格模糊的表述也进行了重新说明,并附上了参考文献来源。

5、药品杂质分析指导原则
该指导原则对于杂质分析具有重要指导意义,里面一大特点是增加了多处关于ICH系列指导原则,如ICHQ3A、ICHQ3B、ICHQ3C、ICHQ3D、ICHQ2等内容,说明ICH指导原则在国内药品研发中已经成为主要的参考指导原则,在药品注册审评过程中也必然会成分主要依据,加强这方面的学习和理解非常必要。

6、分析方法确认指导原则、分析方法转移指导原则
这两个指导原则是2020版药典中新增的指导原则,应该仔细阅读。


关键词:医药化学,医药合成
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2024 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1

